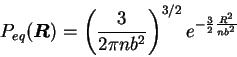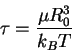A polymeric molecule consists in a long chain formed by the repetition
of a large number of single identical units, the monomers,
linked by chemical bonds.
For typical polymer used in drag reductions
experiments the number of monomers is very large,
![]() and the polymer can be considered, following Kuhn,
as a freely jointed chain on
and the polymer can be considered, following Kuhn,
as a freely jointed chain on ![]() segments of length
segments of length ![]() ,
with independent relative orientation.
,
with independent relative orientation.
When a polymer molecule is put into an homogeneous flow, it assumes the aspect of a statistically spherical coil, because of the thermal agitation.
The average size of the coil, which is also called radius of gyration, can be estimate as the length of the random walk formed by the
On the contrary, in a inhomogeneous flow the molecule
is stretched into an elongated shape, that can be characterized by its
end-to-end distance ![]() , which can be significantly larger than
, which can be significantly larger than ![]() .
The deformation of the molecules is the result of the
competition between the stirring exercised by the gradients of velocity,
and the relaxation of the polymer to its equilibrium configuration,
as a result of Brownian bombardment.
Experiments with DNA molecules [46,47] show
that this relaxation is linear provided that the elongation is smaller
compared to the maximum extension
.
The deformation of the molecules is the result of the
competition between the stirring exercised by the gradients of velocity,
and the relaxation of the polymer to its equilibrium configuration,
as a result of Brownian bombardment.
Experiments with DNA molecules [46,47] show
that this relaxation is linear provided that the elongation is smaller
compared to the maximum extension ![]() (see Fig. 3.1).
(see Fig. 3.1).
![\includegraphics[draft=false, scale=0.7]{P_rilaxpoli.eps}](img455.png)
|
This is consistent with the freely jointed chain model,
where the equilibrium distribution for the end-to-end vector ![]() resulting from the Brownian motion of the
resulting from the Brownian motion of the ![]() elements of the chain
has a Gaussian core:
elements of the chain
has a Gaussian core:
A convenient measure on the relaxation time for the linear chain
is that introduced by Zimm [49]:
Indeed the relaxation process can be much more complex that the simple description given by Zimm model. Several microscopic model of the behavior of polymer molecule has been developed to characterize this process, from the Rouse chain to the Reptation model. An introduction to these models can be found in Doi & Edwards [50]. Nevertheless the simple linear relaxation is able to grasp, at least qualitatively, the basic features of polymer dynamics and feedback.
The relative strength between the relaxation of the polymer and
stretching exerted by the flow is measured by the
Weissenberg number ![]() , defined as the product of the characteristic
velocity gradient and
, defined as the product of the characteristic
velocity gradient and ![]() . When
. When ![]() relaxation is fast
compared with the stretching time, and the polymers remain in their coiled
state. On the contrary, for
relaxation is fast
compared with the stretching time, and the polymers remain in their coiled
state. On the contrary, for ![]() the polymers are stretched
by the flow, and they became substantially elongated.
This transition is known as the coil-stretch transition,
and has been demonstrated to occur under general conditions
in unsteady flow[51,52]
For the case of steady flow the transition is always
present for purely elongational flow, while can be suppressed by rotation,
because the polymers does not point always in the
stretching direction[53].
the polymers are stretched
by the flow, and they became substantially elongated.
This transition is known as the coil-stretch transition,
and has been demonstrated to occur under general conditions
in unsteady flow[51,52]
For the case of steady flow the transition is always
present for purely elongational flow, while can be suppressed by rotation,
because the polymers does not point always in the
stretching direction[53].
In the case of turbulent flows polymers are stretched
by a chaotic smooth flow,
because their size is typically smaller than the viscous
Kolmogorov scale of the fluid.
The intensity of the stretching due to the
gradients of a chaotic smooth velocity field
can be measured by means of the Lyapunov exponent
of Lagrangian trajectories ![]() that is the average logarithmic divergence rate of
nearby fluid trajectories.
The Weissenberg number for chaotic
smooth flow thus reads:
that is the average logarithmic divergence rate of
nearby fluid trajectories.
The Weissenberg number for chaotic
smooth flow thus reads:
The stretching of polymers is limited by their back reaction on the fluid. Indeed the stress tensor for a viscoelastic solution has an elastic component which is proportional to the polymer deformation tensor. When polymer are substantially elongated the elastic stresses can become of the same order of the viscous stresses, and consequently polymers can modify the flow reducing the stretching and giving rise to a dynamical equilibrium state characterized by constant average elongation, which depends on the polymer concentration.
The reduction of the stretching
due to polymers back reaction correspond to a strong
reduction of the Lagrangian Lyapunov exponent of the
viscoelastic fluid[54,55],
thus for the sake of clearness we will always define the
![]() number a-priori as the product of the polymer
relaxation time and the Lyapunov exponent of the
Newtonian fluid
number a-priori as the product of the polymer
relaxation time and the Lyapunov exponent of the
Newtonian fluid
![]() .
.
![\includegraphics[draft=false,scale=0.6]{P_coil.eps}](img446.png)